 ¿Qué es la enfermedad de mucopolisacaridosis tipo II (MPS tipo II)?
¿Qué es la enfermedad de mucopolisacaridosis tipo II (MPS tipo II)?

El síndrome de Hunter o mucopolisacaridosis tipo II (MPS II) es una enfermedad genética hereditaria, ligada al cromosoma X, de la clase de las enfermedades de depósito lisosomal (EDLs). Afecta principalmente a los varones. Existen en el mundo muy pocos casos de mujeres que manifiestan la enfermedad. El síndrome de Hunter es causado por la deficiencia de una enzima llamada iduronato-2-sulfatasa.
La enfermedad de MPS tipo II también es conocida como Síndrome de Hunter. Se nota en los pacientes la ausencia o insuficiencia de enzimas responsables por la descomposición de los mucopolisacáridos, de allí proviene el nombre mucopolisacaridosis. En el caso de la mucopolisacaridosis tipo II, por estar ligada al cromosoma X, el síndrome alcanza —sobre todo— a los hombres y muy raramente a las mujeres. La incidencia es de 1 en cada 155 mil nacimientos. En el mundo, se estima que fueron diagnosticados cerca de 2 mil pacientes. Solo en EE.UU. son 500 los pacientes que padecen el síndrome de Hunter.
Historia de la enfermedad
La MPS fue descripta por primera vez en la historia, por el médico canadiense Dr. Charles Hunter en 1917.
Signos y síntomas principales
La enfermedad interfiere en la capacidad del organismo para descomponer y reciclar determinadas sustancias conocidas como mucopolisacáridos, o glicosaminoglicanos (GAG) en el lisosoma, que da por resultado una disfunción orgánica multisistémica. A medida que los GAG se acumulan en las células de todo el cuerpo, las señales del síndrome de Hunter se vuelven más visibles. Las manifestaciones son diversas, como alteraciones faciales, cabeza con volumen mayor, abdomen aumentado, pérdida auditiva, compromiso de las válvulas del corazón, que lleva a una declinación de la función cardíaca, obstrucción de las vías respiratorias, apnea del sueño, aumento del hígado y del bazo.
Puede, además, afectar la movilidad por la acumulación de GAG en las articulaciones (las «coyunturas»). En algunos casos, el sistema nervioso central puede resultar comprometido. No todas las personas portadoras del síndrome son afectadas de la misma manera. Sin embargo, la enfermedad es siempre grave, progresiva, crónica y, si no se diagnostica y trata a tiempo, puede ocasionar la muerte. Vale recordar que en el caso de MPS II el niño se presenta normal al nacer. Sin embargo, en los dos primeros años de vida, la progresión de la enfermedad es muy rápida. Se estima que el tiempo para el diagnóstico desde la aparición de los primeros signos y síntomas es de 7 años. Si la enfermedad no se diagnostica y se trata precozmente, la expectativa de vida para los portadores del síndrome es de aproximadamente 15 años.
¿Quién puede contraer la enfermedad de MPS tipo II?
La enfermedad MPS tipo I es heredada, es decir, causada por genes mutantes o defectuosos que son transmitidos por los padres a los hijos.
Diagnóstico
Actualmente, el principal obstáculo para el diagnóstico y tratamiento de pacientes portadores del síndrome de Hunter y de enfermedades genéticas raras, es el desconocimiento de los médicos y profesionales de la salud en general, que trae como consecuencia un diagnóstico tardío y, muchas veces, erróneo de la enfermedad. Esto se debe al hecho de que los síntomas presentados por los pacientes son fácilmente confundidos con enfermedades comunes en niños.
Existen dos métodos disponibles para diagnosticar el síndrome de Hunter. El primero y más utilizado es un examen de orina, para investigar los niveles de los glicosaminoglicanos (GAG). Éste es el primer paso en la investigación de la enfermedad. La confirmación exacta del síndrome se hace solamente con una prueba para medir la actividad enzimática de la sangre o de la piel del paciente.
La genética de MPS tipo II
Casi todas las células del cuerpo humano tienen 46 cromosomas, siendo 23 derivados de cada uno de los padres. El gen que codifica la producción del I2S está localizado en el cromosoma X. Las personas del sexo femenino tienen dos cromosomas X, uno heredado del padre y otro de la madre; las personas del sexo masculino tienen un cromosoma X heredado de la madre y un cromosoma Y heredado del padre. Si un individuo tiene la copia anormal del gen para I2S desarrollará el síndrome de Hunter.
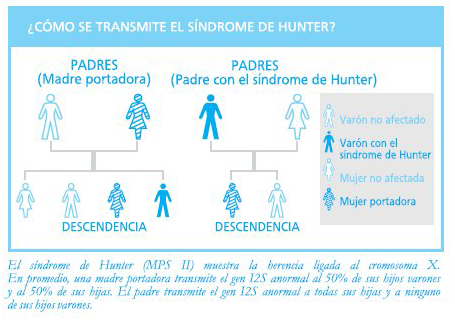
El síndrome de Hunter posee un patrón de herencia ligado al cromosoma X. La madre portadora trasmitirá el gen con la mutación codificadora I2S, con una probabilidad de 50% en cada gestación. El padre con síndrome de Hunter trasmitirá el gen con la mutación a todas sus hijas, pero no lo trasmitirá a ninguno de sus hijos.
La bioquímica de MPS tipo II
El cuerpo humano depende de una amplia gama de reacciones bioquímicas para mantener sus funciones vitales, incluso para producir energía, crecer y desarrollarse, para las comunicaciones dentro del cuerpo y para protección contra infecciones. Otra función crítica es la descomposición de biomoléculas grandes. Es ahí donde reside la base del problema del síndrome de Hunter (MPS II) y de otras enfermedades de depósito lisosomal.
La bioquímica del síndrome de Hunter está ligada a un problema en una parte del tejido conjuntivo conocido como matriz extracelular. La matriz está constituida por varios azúcares y proteínas y ayuda a formar la estructura arquitectónica de soporte del organismo. La matriz circunda las células del organismo como una malla organizada, y funciona como una pegatina que mantiene las células del organismo unidas. Uno de los componentes de la matriz extracelular es una molécula denominada proteoglicano. Como muchos componentes del cuerpo, los proteoglicanos necesitan ser descompuestos y sustituidos. Cuando el organismo descompone los proteoglicanos, uno de los productos resultantes son los mucopolisacáridos, también conocidos como GAG. Hay varios tipos de GAG, cada uno localizado en determinados lugares del organismo.
En el síndrome de Hunter, el problema reside en la descomposición de dos GAG: el dermatán sulfato y el heparán sulfato. La primera etapa de la descomposición del dermatán sulfato y del heparán sulfato requiere la enzima lisosómica I2S. En personas que sufren el síndrome de Hunter, esa enzima está parcial o completamente inactiva. Como consecuencia, los GAG se acumulan en las células de todo el cuerpo, en particular en los tejidos que contienen dermatán y heparán sulfato. Como consecuencia de esa acumulación ocurren interferencias en el modo de funcionamiento de determinadas células y órganos, lo que ocasiona una serie de síntomas graves. La tasa de acumulación de GAG no es la misma para todas las personas con el síndrome de Hunter, lo que conduce a la aparición de una serie de problemas médicos.
| GAG | Localización corporal |
| Ácido hialurónico | Varios tejidos conectivos, piel, cartílagos, líquido sinovial |
| Condroitin sulfato | Cartílagos, córnea, huesos, pies, arterias |
| Dermatán sulfato | Piel, vasos sanguíneos, corazón, válvulas cardiacas |
| Heparán sulfato | Pulmones, arterias, superficie celular |
| Heparina | Pulmones, hígado, ciertas células del sistema inmunológico |
| Keratán sulfato | Cartílagos, cornea, discos intervertebrales |
Tratamiento
Se realizaron varios intentos para ayudar al organismo a producir enzimas I2S normales. Esos intentos incluyeron la transferencia de células de la sangre, de membranas amnióticas, de la médula ósea, y trasplante de sangre del cordón umbilical de individuos no afectados hacia portadores del síndrome de Hunter. Sin embargo no fue posible comprobar la mejoría en el estado clínico de los pacientes a largo plazo. A pesar de que el trasplante de células de la médula ósea en pacientes con formas graves de MPS tipo I tuvo un relativo grado de éxito cuando se realizó precozmente, en MPS II la técnica no presentó resultados satisfactorios, siendo, por lo tanto, no indicada rutinariamente. Pueden resultar útiles terapias de soporte, incluyendo fonoaudiología, fisioterapia y ciertos procedimientos quirúrgicos.
La terapia que trata la causa de la enfermedad, la falta de la enzima I2S, está aprobada en diversos países. La terapia de reemplazo enzimática (TRE) es un abordaje para el tratamiento del síndrome de Hunter, que implica la reposición de la enzima deficiente por infusiones intravenosas en personas aquejadas por la enfermedad. Varios estudios demostraron que los pacientes tratados con esta enzima presentan una mejora significativa del cuadro clínico de la enfermedad.