¿Qué es la enfermedad de LAL-D?
La deficiencia de LAL es una enfermedad grave y potencialmente fatal, para la cual no hay aún tratamientos aprobados.
La deficiencia de lipasa ácida lisosomal (LAL) es parte de una familia de afecciones llamadas enfermedades por depósito lisosomal. Los lisosomas se encuentran en las células del cuerpo y desempeñan un papel importante en la digestión de nutrientes y otros materiales. En condiciones normales, el cuerpo produce una enzima llamada lipasa ácida lisosomal (LAL). La enzima LAL degrada material graso (ésteres de colesterol y triglicéridos). La deficiencia de LAL ocurre cuando el cuerpo no produce esa enzima en cantidad suficiente.
Los bebés, niños y adultos que sufren de deficiencia de LAL experimentan una serie de problemas de salud muy graves y devastadores. La falta de la enzima LAL resulta en una acumulación de material graso en el hígado, el bazo y los vasos sanguíneos. En algunos pacientes, esta acumulación también se produce en el intestino y otros órganos o áreas importantes del cuerpo.
La deficiencia de LAL que se manifiesta en los bebés, a veces conocida como enfermedad de Wolman (por Moshe Wolman, el médico que la describió por primera vez), es casi universalmente fatal en los primeros seis meses de vida.
La deficiencia de LAL en niños y adultos es también llamada enfermedad por depósito de ésteres de colesterol (EDEC, o CESD en inglés). Los pacientes sufren una acumulación de material graso, sobre todo en el hígado y las paredes de los vasos sanguíneos, aunque otros órganos del cuerpo pueden verse igualmente afectados. El depósito de material graso en las células del hígado puede provocar hepatomegalia (agrandamiento del hígado), cirrosis e insuficiencia hepática crónica.
¿Con qué otros nombres se conoce LAL-D?
La deficiencia de LAL es también llamada de:
- Deficiencia de hidrolasa ácida de ésteres de colesterol, tipo 2
- Carencia de lipasa ácida
- Enfermedad por depósito de hidrolasa de ésteres de colesterol
- Enfermedad por depósito de ésteres de colesterol (EDEC, o en inglés CESD)
- Enfermedad de Wolman
Otros trastornos relacionados
- Esteatohepatitis no alcohólica (EHNA, o en inglés NAFDL)
- Enfermedad hepática alcohólica (en inglés NASH)
- Cirrosis criptogénica
- Enfermedad de Niemann-Pick tipo C
- Síndrome de Chanarin-Dorfman
¿Cómo se contrae la enfermedad LAL-D?
La deficiencia de LAL es un trastorno heredado. El gen responsable de decirle al cuerpo cómo producir una enzima LAL es anormal, entonces esta enzima o bien no funciona correctamente o no es producida en absoluto. Cada persona hereda dos copias del gen de la LAL: una copia de su madre y otra de su padre.Una persona con deficiencia de LAL ha heredado dos copias defectuosas de este gen, una de cada progenitor. Si alguien tiene una sola copia del gen defectuoso, no suele desarrollar la enfermedad, aunque la persona en esta situación se convierte en un portador de la enfermedad y puede pasar la copia defectuosa a sus hijos. Cuando ambos padres son portadores, existe un 25% de probabilidad en cada embarazo (uno de cada cuatro) de que su hijo tenga la deficiencia de LAL.
¿Cómo se diagnostica la deficiencia de LAL?
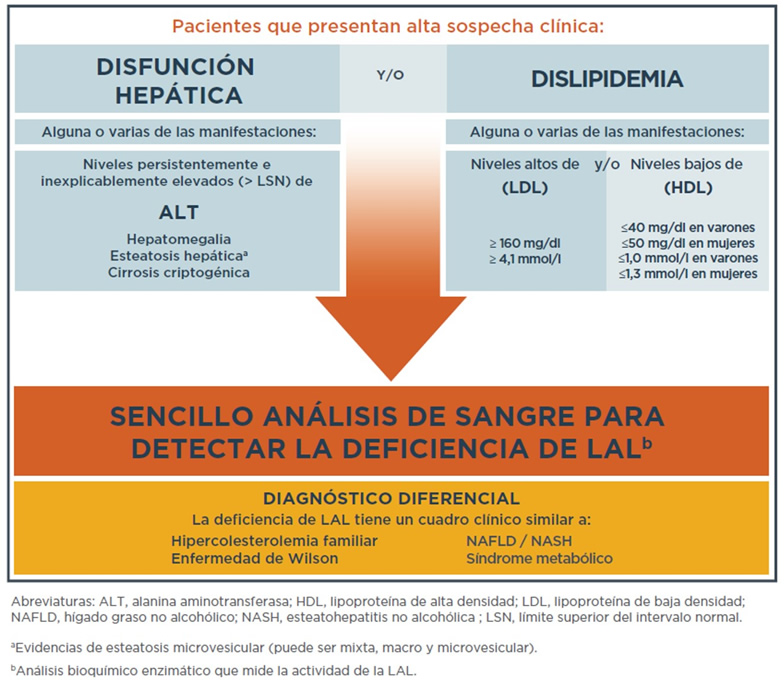
Las posibles anomalías que hacen que un médico considere la deficiencia de LAL incluyen:
- Aumento de las enzimas hepáticas (transaminasas: AST y ALT)
- Alto nivel de colesterol y triglicéridos
- Alto nivel de colesterol “malo” (LDL)
- Nivel muy bajo de colesterol “bueno” (HDL)
- Hígado agrandado debido al depósito de grasa
A menudo, los médicos no reconocen la deficiencia de LAL porque no han tenido experiencia con este trastorno. El diagnóstico comienza con una consulta y la elaboración de la historia clínica, seguidas por un examen y pedido de pruebas laboratoriales preliminares. El médico puede investigar o preguntar sobre los siguientes problemas hepáticos:
- Hepatomegalia (agrandamiento del hígado) inexplicable
- Enzimas hepáticas elevadas (un marcador del daño al hígado)
- Material graso o lipídico inexplicable en el hígado
- Enfermedad hepática crónica inexplicable que puede estar empeorando con el tiempo
Algunas enfermedades comunes que se asemejan a la deficiencia de LAL y pueden conducir a un diagnóstico equivocado son:
- Causas infecciosas, como la hepatitis viral
- Causas metabólicas, como la enfermedad del hígado graso no alcohólico (EHGNA) o la esteatohepatitis no alcohólica (EHNA)
- Enfermedad hepática autoinmune (cuando el sistema inmunológico del cuerpo ataca a las células del hígado)
Otras pistas que pueden ayudar al médico a diagnosticar la deficiencia de LAL son:
- Niveles bajos de HDL, especialmente si son muy bajos
- Baja estatura
- Ganglios linfáticos agrandados
- Bazo agrandado
Estas características son menos comunes en los pacientes con otras enfermedades hepáticas y, por lo tanto, son pistas importantes que pueden indicar al médico el diagnóstico de deficiencia de LAL.
Una vez que el médico considere la deficiencia de LAL como causa posible de determinados problemas de salud, puede solicitar una prueba confirmatoria, como un análisis enzimático (leucocitos o muestra de sangre seca) o un análisis de secuenciación genética. El análisis enzimático mide el nivel y la actividad de la enzima LAL, y la secuenciación genética ayuda a identificar si alguien tiene una mutación genética compatible con la deficiencia de LAL. Esta enfermedad es progresiva y resulta en daños al hígado, al bazo y a otros órganos internos, además de poder provocar fibrosis, cirrosis, insuficiencia hepática y muerte. Un diagnóstico preciso es el primer paso para tratar las complicaciones a largo plazo graves y, a menudo, fatales de esta enfermedad.
Signos y valores de laboratorio que deben levantar la sospecha de la deficiencia de LAL
¿Cómo se trata la deficiencia de LAL?
No hay tratamientos aprobados para la deficiencia de LAL. Sin embargo, se está estudiando el uso de sebelipasa alfa como terapia de reemplazo enzimático para esta enfermedad. Las terapias de reemplazo enzimático han sido utilizadas con éxito para el tratamiento de otras enfermedades por depósito lisosomal.
A los niños y adultos con deficiencia de LAL, el médico puede recetar fármacos para reducir el colesterol (estatinas o ezetimiba), para tratar los altos niveles de colesterol y otras grasas en la sangre (alto LDL o colesterol “malo”). Aunque estos fármacos pueden, en cierta medida, reducir los niveles séricos de colesterol, no han demostrado mejorar la enfermedad subyacente y los problemas hepáticos graves. Además, hay evidencias de que estos fármacos, en realidad, pueden ocultar el avance de la enfermedad.